


文摘
越来越多的证据牵连了降钙素相关基因肽(CGRP怎样)受体在偏头痛病理生理学。批准单克隆抗体针对CGRP怎样或CGRP怎样受体,抑制CGRP-mediated信号成为一种很有前途的方法成人偏头痛的预防治疗。最近,小分子anti-CGRP治疗显示治疗偏头痛的疗效。目前的研究旨在描述ubrogepant的药理学特性,一个口头可利用CGRP怎样受体拮抗剂对急性治疗偏头痛。在一系列的配体结合化验,ubrogepant展出本土的高亲和力(K我= 0.067 nM)和克隆人类(K我= 0.070 nM)和恒河CGRP怎样受体(K我= 0.079海里),相对较低的亲和力CGRP怎样受体的老鼠,老鼠,兔子和狗。在功能分析,ubrogepant强有力地阻止了人类α营地cgrp怎样−刺激响应(IC500.08 nM)和CGRP怎样表现出高度选择性拮抗剂活性受体与其他人类降钙素受体家族的成员。此外,体内CGRP怎样受体拮抗剂ubrogepant活动在药效学评价模型capsaicin-induced真皮血管舒张(CIDV)在恒河猴和人类。结果表明,ubrogepant产生浓度抑制CIDV意味着电子商务503.2和2.6 nM的恒河猴和人类,分别。猴子的大脑渗透研究ubrogepant显示脑脊液:等离子体的比率0.03,CGRP怎样受体入住率低。总之,ubrogepant亲和力高的竞争对手,力量,人类CGRP怎样受体的选择性。
意义的声明Ubrogepant是一个强有力的,有选择性的,口头传递,小分子人类CGRP怎样的竞争对手。体内研究使用药效学模型CIDV的恒河猴和人类证明ubrogepant CIDV浓度产生抑制,表明预测pharmacokinetic-pharmacodynamic关系。
介绍
偏头痛是一种非常普遍、慢性神经系统疾病和残疾的主要原因在15 - 49岁的人(伯奇et al ., 2018;施泰纳et al ., 2018)。常用的急性偏头痛发作的治疗方法包括药物,阿片类药物,非甾体类抗炎药物,麦角胺衍生品,巴比妥酸盐和组合止痛剂(荷兰et al ., 2013;Martelletti 2017)。但是,这些治疗的效用受到低水平的依从性和病人满意度主要源自不足疗效和耐受性差(荷兰et al ., 2013;立顿et al ., 2013;Messali et al ., 2014;塞拉诺et al ., 2015;Martelletti 2017)。因此,许多偏头痛患者停止急性偏头痛治疗和可能会经历不受控制的攻击或疾病进展(荷兰et al ., 2013;2016年5月,舒尔特吗;Thorlund et al ., 2016)。
理论解释偏头痛的病理生理学已经从一个纯粹的转移向神经源性血管疾病模型理论关注神经肽降钙素相关基因肽(CGRP怎样)(汉弗莱,2007;Eftekhari Edvinsson, 2010;摩尔和萨尔瓦多,2012年;Gonzalez-Hernandez et al ., 2018)。CGRP怎样37-amino酸肽和强有力的血管舒张药现在在高位trigeminovascular系统在偏头痛发作(戈德比et al ., 1990;罗素et al ., 2014)。肽的降钙素家族包括降钙素(CT)、糊精(艾米)adrenomedullin (AM),和CGRP怎样(Poyner et al ., 2002)。CGRP怎样受体由CT受体(CTR)——受体(CLR)和受体活性修饰蛋白1 (RAMP1) (Eftekhari Edvinsson, 2010;Kiriyama Nochi, 2018)。分布的CLR和RAMP1映射到三叉神经元的胞浆,在外围网站颅内血管平滑肌细胞(),在硬脑膜(血管和缺血性本地化),并在脑干(Edvinsson Warfvinge, 2019)。CTR的艾米受体是一个复杂和RAMP1(即。,艾米1),RAMP2(即。,艾米2),或者RAMP3(即。,艾米3艾米有一个高度的亲和力。艾米的1受体结合CGRP怎样,在三叉神经节中找到,并已与疼痛信号的动物模型;然而,它在偏头痛病理生理学中的作用仍有待决定(Gebre-Medhin et al ., 1998;Poyner et al ., 2002;沃克et al ., 2015;Kiriyama Nochi, 2018;Edvinsson Warfvinge, 2019)。是受体是一个复杂的CLR和RAMP2 (1)和RAMP3 (2点(),它有一个高度的亲和力Kiriyama Nochi, 2018)。受体在偏头痛的功能是未知的,而且,与CGRP怎样,静脉输液是不存在沉淀偏头痛(彼得森et al ., 2009)。
大量的科学数据引起CGRP怎样通路在底层与偏头痛相关的生理机制(戈德比et al ., 1990;拉森et al ., 2002;Edvinsson 2015)。此外,先前研究小分子CGRP怎样受体拮抗剂显示功效治疗偏头痛,尽管发展最终停止了出于安全考虑(休伊特et al ., 2011;何鸿燊et al ., 2014;哈格里夫斯和奥尔森,2019)。单克隆抗体的临床成功针对CGRP怎样还提供了支持CGRP怎样受体作为一个有前途的治疗偏头痛的目标(Dodick 2019)。
Ubrogepant是一种口服生物利用率、强有力的和具体的CGRP怎样受体拮抗剂,2019年12月被批准为偏头痛的急性治疗成人有或没有气场。Ubrogepant是化学不同于以前的小分子CGRP怎样受体拮抗剂(Ubrelvy 2019)(Yasuda et al ., 2017)。在临床试验中,ubrogepant提供实质性缓解疼痛,恢复功能,一般耐受性良好。目前沟通的目的是描述ubrogepant药理学的状况。
材料和方法
体外药理学
绑定关联。
评估ubrogepant CGRP怎样,受体的亲和力和选择性,克隆CGRP怎样和我2受体稳定表达人类胚胎kidney-derived (HEK293)细胞。评估ubrogepant艾米的亲和力1克隆受体,艾米1受体被转染瞬变猴子kidney-derived细胞表达与等量的CTR RAMP1监控ubrogepant这个受体的选择性。
膜受体结合化验,分数被孤立的从HEK293细胞匀浆或猴子kidney-derived细胞或匀浆的小脑恒河隔绝,老鼠,老鼠,兔子和狗。人类(125年我)CGRP怎样和125年我]和老鼠125年我]艾米被用作放射性配体结合化验。非特异性结合是由10µmk - 3207 (萨尔瓦多et al ., 2010),一个结构不同的CGRP怎样受体拮抗剂。1毫升的测试进行了绑定缓冲(10毫米玫瑰,pH值7.4,5毫米MgCl2,0.2%的牛血清白蛋白)3小时在室温下人类(包含10点125年晚上10点我)CGRP怎样,人类(125年我),或40点鼠(125年我]艾米的几个ubrogepant浓度。化验是终止通过过滤0.5% polyethyleneimine-treated GF / B与冰冷的玻璃纤维过滤板清洗缓冲(10毫米玫瑰,pH值7.4和5毫米MgCl2)。闪烁液添加到盘子和放射性量化使用帕卡德TopCount NXT闪烁计数器(PerkinElmer,谢尔顿,CT)。剂量反应曲线被绘制确定half-maximal抑制浓度(IC50)值和转换为K我值使用方程K我=集成电路50(配体)/ l + () /Kd)。数据组织方式和S.E.M.通过叙述地呈现出来年代,除非另有注明。
功能的能力。
ubrogepant CGRP怎样——的影响,AM -,或CT-induced增加环腺苷酸(营)是人类CGRP怎样评估在HEK293细胞中表达受体,恒河CGRP怎样受体,克隆人类1(CLR / RAMP2)2(CLR / RAMP3),克隆人类的艾米1(CTR / RAMP1)或艾米3(CTR / RAMP3),或人类CTR孤单。
细胞pre-incubated在384 - 2000细胞/孔板与不同浓度的ubrogepant 30分钟37°C。在人类CGRP怎样受体功能化验、效能评估,没有50%的人类和恒河猴血清。环核苷酸抑制剂isobutyl-methylxanthine被加入到细胞浓度达到300µ米30分钟37°C刺激为1.0 nM人类紧随其后αCGRP怎样(人类和恒河CGRP怎样受体分析)、1.0 nM人类是人类1,我2受体分析)、0.5 nM鼠艾米艾米(克隆人类1和艾米3人类受体分析),或0.2 nM CT(人类CTR化验)20分钟37°C。受体激动剂刺激后,营地浓度与均相时间分辨荧光测量营地动态分析(Cisbio,贝德福德,MA)。
剂量反应曲线绘制和集成电路50从四个参数值确定物流符合定义的方程y = [(−d) / (1 + x / c)b)+ d, y =反应剂量x =, =最大响应,d =最低响应,c =拐点,b =斜率。Schild CGRP怎样化验,分析被用作衡量竞争对抗的策划(DR-1)和[B],对数博士的比例αcgrp怎样half-maximal有效浓度(EC50)值的存在和缺乏ubrogepant和(B)拮抗剂的浓度。x轴截距等于巴勒斯坦权力机构2和KB使用公式计算2=−日志KB。
专一性/非目标分析。
ubrogepant是评估的特异性配体结合跨116年的目标或功能分析(补充表1;Olon Ricerca生物科学、和谐、OH)和人类ether-a-go-go-related基因(hERG),编码inward-rectifying电压门控钾通道在心脏和心脏复极化。Ubrogepant 10浓度进行了测试µ常规放射性配体结合,酶化验,浓度和剂量反应曲线生成重大活动时被观察到。
的hERG配体结合试验进行了使用膜分数隔绝HEK293细胞稳定表达克隆hERG。(35S] mk - 499 (王et al ., 2003)是用作放射性配体和10µ阿司咪唑(Suessbrich et al ., 1996)是用来确定非特异性结合。绑定进行了化验1毫升的绑定缓冲(60 mM氯化钾、氯化钠71.5毫米,1毫米CaCl22毫米MgCl2,玫瑰和10毫米,pH值7.4)包含25点(35S] hERG配体浓度的几个ubrogepant 3小时在室温下。试验终止通过过滤0.05% polyethyleneimine-treated GF / B与冰冷的玻璃纤维盘子洗缓冲区(10毫米玫瑰,pH值7.4)。盘子在37°C真空下干燥1小时,闪烁液补充说,放射性量化使用帕卡德TopCount NXT闪烁计数器。
绑定动力学。
饱和结合分析进行结合浓度增加(3H] -ubrogepant 10µCGRP怎样受体拮抗剂mk - 3207为非特异性结合,和50µg SK-N-MC膜/。一夜之间,混合物被孵化(18小时)在室温下在绑定缓冲(10毫米玫瑰,pH值7.4,5毫米MgCl2,0.2%的牛血清白蛋白)总量的1毫升。
协会进行了动力学分析相结合(40点3H] -ubrogepant 50µ每在绑定缓冲和g SK-N-MC膜在室温下孵化为不同时期(1 - 90分钟)。解离动力学分析相结合进行40点(3H] -ubrogepant 50µ每在绑定缓冲和g SK-N-MC膜在室温下孵化为3小时。在这一点上,10µCGRP怎样的M受体拮抗剂mk - 3207添加和离解监控各种时间间隔(1 - 300分钟)。所有化验终止通过过滤0.5% polyethyleneimine-treated GF / B与冰冷的玻璃纤维盘子洗缓冲(10毫米玫瑰,pH值7.4,5毫米MgCl2)。盘子在37°C真空下干燥1小时,闪烁液补充说,放射性量化使用帕卡德TopCount NXT闪烁计数器。
体内药理学:评估的药效学作用
CGRP怎样的药效学(PD)活动受体拮抗剂体内已经建立和验证使用capsaicin-induced真皮血管舒张(CIDV)在恒河猴模型(萨尔瓦多et al ., 2008,2010年)和人体临床试验(李et al ., 2015)。因此,体内PD活性ubrogepant评估使用CIDV试验在恒河猴和人类在这个研究。人类的协议CIDV研究是审查和批准的独立鲁汶大学医院的伦理委员会,比利时。在入学之前,所有的参与者给书面知情同意后,一个完整的口头和书面的解释研究。这项研究是依照当地法律进行赫尔辛基宣言的道德原则,和良好的临床实践指南。
恒河猴CIDV。
21岁成年恒河猴(≤10 /自习)被使用在6个研究来确定车辆的影响和积极CIDV上测试代理。动物提供至少5 - 7 procedure-free天之间的研究。CIDV测试、动物保持在异氟烷麻醉,和四个o型环(内部直径8毫米)放在腹侧前臂。平衡后,2毫克的基线反应20分钟后应用辣椒素(Tween20乙醇溶解在30%,30%,40%水)在一个环测量用激光多普勒成像(沼泽仪器,威尔明顿,德)。
接下来,连续三静脉丸+静脉输液的车辆或一至三个剂量的上升ubrogepant接种。每个输液开始后的五分钟,2毫克辣椒素应用于一个剩余的戒指。扫描完成后每个环开始之前的辣椒素的应用程序(即后浸泡20分钟。25分钟后,开始注入)。研究目标ubrogepant等离子体水平的0.5,5日和50 nM五男两女恒河猴;研究B有针对性的等离子体水平的5,50和150 nM六男一女恒河猴;研究C有针对性的等离子体水平的1、5、10 nM的三个雌性恒河猴;研究D有针对性的等离子体水平的150、500和500海里七雄性恒河猴;研究E有针对性的等离子体水平的1 10和10 nM两男两女恒河猴;F和研究等离子体水平的目标在10雄性恒河猴400海里。剂量校准实现指定目标离子水平和提供足够的保险动态范围的药代动力学(PK) / PD曲线。
血液样本,以确定等离子体ubrogepant浓度,得到了响应曲线和抑制浓度在20分钟后应用程序的每个测试期间。实证最大效应(E马克斯)模型是用来描述的PK / PD关系ubrogepant CIDV抑制的恒河猴。血液流动被形容为一个基线血流+增量血流由于CIDV和封锁CIDV ubrogepant通过E马克斯的关系。该模型表示为F = F0+ F帽•[1−E马克斯•C / (EC50+ C)], F是观察到的血流量(意思是灌注值)来衡量激光多普勒成像,F0基线血液流动,F帽的增量血流由于应用辣椒素,E马克斯最大比例的抑制ubrogepant C ubrogepant的血浆浓度,和电子商务50的血浆浓度抑制CIDV ubrogepant对应于50%。数据集中在6个恒河CIDV研究(静脉注射剂量范围从0.06到100μ克/公斤)(见补充表2剂量为每一个研究)。个人间的变化参数选择使用向前替换为F(显著性水平为0.05)0F帽E马克斯,电子商务50。协变量的评估侧重于寻找研究中差异F0辣椒素(F和响应帽)。模型拟合进行了使用NONMEM七世(图标plc,都柏林,爱尔兰)使用一阶条件估计与互动。
人类CIDV。
健康的年轻男性18-50岁期间服用口服ubrogepant随机,双盲,安慰剂对照,本研究交叉研究(EudraCT号码:2011-002359-32)。参与者被要求禁食8小时前ubrogepant剂量和预处理程序。抑制CIDV被激光多普勒测量扫描1和5小时后单剂量口服ubrogepant(0.5毫克,5毫克,40毫克)。剂量被选来捕捉我们的预期动态范围曲线基于估计EC503.2 nM恒河CIDV实验。多普勒扫描以前也进行了研究(包含目的)和前研究药物管理局(初始剂量)。辣椒素是前30分钟每个postdose应用激光多普勒扫描在0.5和4.5小时后研究药品监督管理局。辣椒素是应用单一局部剂量的300μg / 20μl和1000μg / 20μl辣椒素在10毫米橡胶o形圈在两个站点的手掌的表面参与者的左和右前臂。数据被用来确定药物的浓度必须达到欧盟90年使用PK / PD模型和使用两个剂量的辣椒素,使用一个方法类似于上述灵长类动物的研究。
大脑渗透研究
CGRP怎样受体Ubrogepant入住率的恒河猴大脑正电子发射断层扫描。
所有动物实验进行了符合指南的护理和使用实验动物(生命科学研究所实验动物资源委员会,国家研究委员会,2011年)和被批准的机构动物保健和使用委员会默克公司Inc .(西点军校,PA)。CGRP怎样受体入住率的量化ubrogepant四麻醉成年雄性猕猴进行了正电子发射断层扫描术(PET),使用的PET示踪CGRP怎样受体(11C] mk - 4232 (Hostetler et al ., 2013)。一个基线进行PET扫描与11C] mk - 4232没有ubrogepant。建立稳定的等离子体水平的研究药物,静脉丸+不断注入ubrogepant开始前60分钟静脉丸注入∼5 mCi (11C] mk - 4232和持续的时间扫描。宠物研究获得120分钟后(11C] mk - 4232管理。
等离子体的浓度(11C] mk - 4232对每个研究获得的总放射性测量动脉血浆,修正的部分完整的示踪剂的高效液相色谱确定,和等离子ubrogepant水平测定动脉血液样本。组织时间曲线符合,受体入住率计算使用拉森阴谋。对于每个receptor-occupancy宠物研究,估计受体入住率与PET扫描期间的平均血浆药物水平。
脑脊液渗透Ubrogepant恒河猴。
脑脊液(CSF):等离子体的比例ubrogepant评估在三个成年雄性猕猴慢性小脑延髓池植入导管和港口系统重复的CSF的集合。其他细节与CSF收集之前发表(吉尔伯托et al ., 2003;萨尔瓦多et al ., 2010)。口服后的ubrogepant填喂法10毫克/公斤,脑脊液和血浆样本收集在基线和0.5,1,2,4,8,24小时和分析化合物的水平。
结果
Ubrogepant是一种口服生物利用率CGRP怎样受体拮抗剂用于偏头痛的急性治疗。的化学结构ubrogepant提出了图1。

ubrogepant的化学结构。
Ubrogepant受体结合和功能的潜力。
Ubrogepant是一个强有力的抑制剂(125年我)CGRP怎样绑定的克隆人类CGRP怎样受体和本地的意思K我(±S.E.M.) 0.07±0.006和0.067±0.004 nM,分别为(表1)。类似的亲和力与恒河CGRP怎样观察ubrogepant受体0.079±0.005 nM;然而,明显低亲和力被发现老鼠,老鼠,兔子和狗受体(K我> 9.5海里)。亲和力的人2受体是显著降低(K我= 2059±122海里)比人类CGRP怎样受体,但ubrogepant显示重组人类艾米的温和的亲和力1通过抑制受体(125年我和一个]-rAMY绑定K我8.2 nM(个人K我= 6.5,9.8)。
Ubrogepant强有力地阻止了人类α-CGRP-stimulated营地反应意味着(±S.E.M.)集成电路500.081海里(0.005海里)在人类CGRP怎样receptor-expressing HEK293细胞和0.07海里(0.02 nM)恒河CGRP怎样receptor-expressing HEK293细胞(表2)。添加50%的人类或50%恒河血清ubrogepant表面的实力降低了大约2.4——4.0倍人类和恒河(0.19±0.01海里)(0.30±0.01海里)CGRP怎样受体,分别。使用席尔德回归,ubrogepant造成的,向右存在剂量依赖的相关性变化(数据未显示)受体激动剂的剂量反应曲线,KB= 0.017海里,没有减少最大受体激动剂的反应。
Ubrogepant的特异性和选择性。
116年对特异性评估酶,受体和离子通道绑定化验(补充表1),ubrogepant显示多巴胺转运体弱关联(K我= 4440海里),远远低于其CGRP怎样受体的亲和力。饱和结合研究使用(3H] -ubrogepant表明特定的绑定是饱和SK-N-MC膜,KD0.041纳米。
Ubrogepant显示没有明显的拮抗作用AM-induced营地刺激人类的1或人类CTR浓度大于20000纳米,而效力是有点大2受体(受体和符合绑定数据表3)。同样,在封锁AMY-stimulated营地反应,ubrogepant证明拮抗剂活动在人类艾米1和艾米3受体在效能与基于[这些受体的亲和力125年我]-rAMY绑定(表3)。
药效学评价。
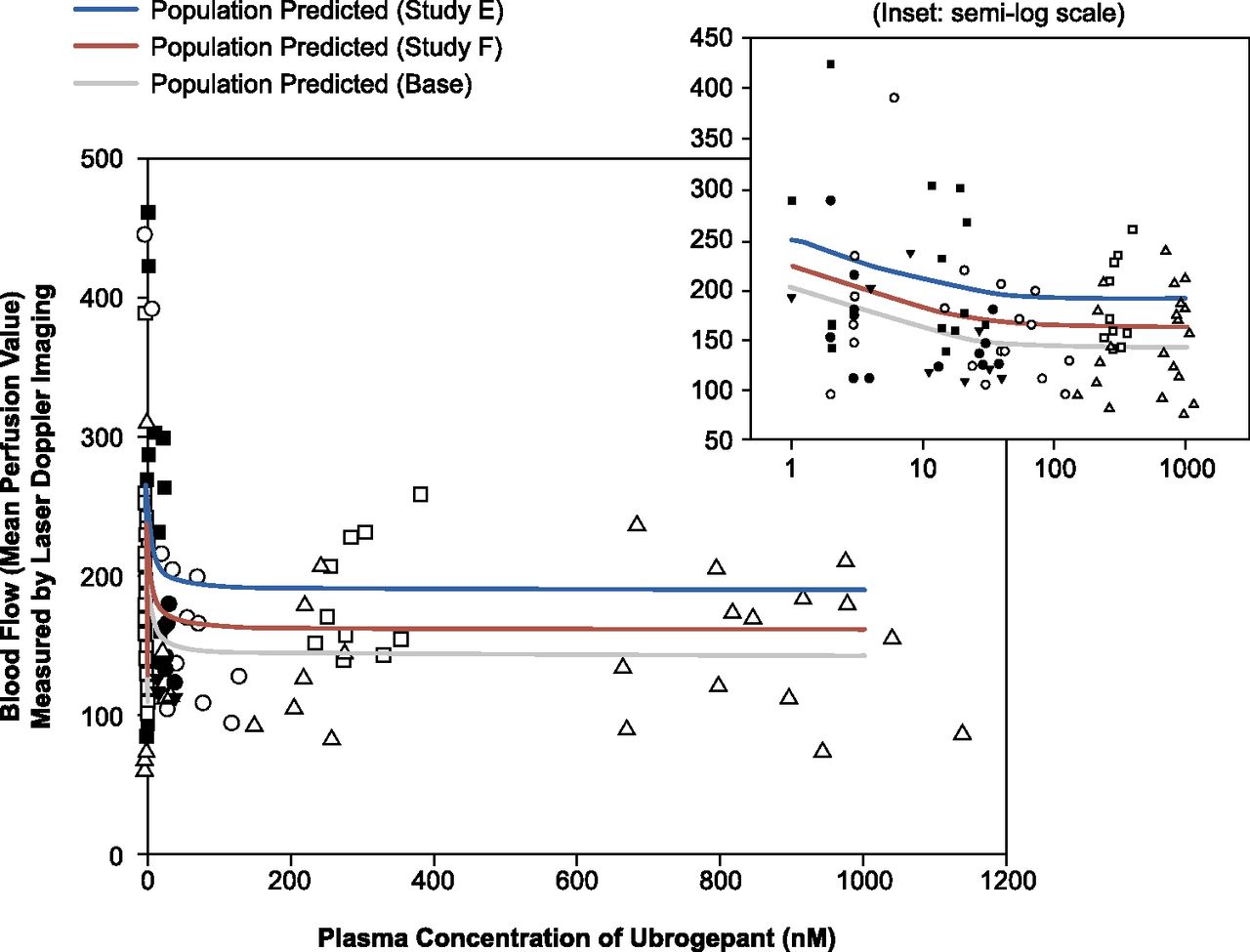
真皮血管舒张反应辣椒素被发现浓度和时间。应用车辆本身没有明显抑制产生的血流量增加。PK / PD关系抑制CIDV ubrogepant估计是基于数据从六个恒河CIDV研究(研究f,静脉注射剂量范围从0.06到100μ使用人口E g / kg)马克斯模型(补充表2)。研究E(0.3和3μ克/公斤)和F (50μ克/公斤)被发现是重要的反是基线血液流动管理辣椒素或ubrogepant之前(即。F0)(图2)。Ubrogepant意味着电子商务503.19 nM (S.E.M.3.65纳米;补充表3估计),相应的电子商务90年29海里。E马克斯为抑制CIDV ubrogepant是0.732 (±0.0859)。

Ubrogepant剂量依赖性抑制capsaicin-induced恒河前臂皮肤血管舒张:人口模型预测和观察血流后2毫克辣椒素应用程序在不同的等离子体浓度Ubrogepant。数据汇集从六个恒河CIDV研究(研究与静脉注射剂量范围从0.06到100 Fμ克/公斤)用符号表示。实线代表的模型预测人口平均值。研究E和F被发现在统计上有显著差异的基线(即血液流量。,before administration of capsaicin or ubrogepant) and thus the model-predicted population means are shown separately.
在人类CIDV PD的研究,单剂量的剂量依赖性降低观察ubrogepant postdose与安慰剂比较1和5小时,无论使用辣椒素浓度(表4)。估计EC50和电子商务90年值ubrogepant CIDV抑制的人2.56和23海里,分别。
讨论
Ubrogepant是一个强有力的CGRP怎样受体拮抗剂用于偏头痛的急性治疗。在目前的研究中,ubrogepant展出人类CGRP怎样的高亲和力受体(K我0.07海里)。此外,ubrogepant展出物种特异性,显示人类和恒河CGRP怎样高亲和力受体,减少为其他非人类受体亲和力。
Ubrogepant显示人工CGRP怎样受体高选择性和人类1,是2、CT和艾米3但选择性受体减少对艾米1受体。这个观察是一致的其他小分子的RAMP1-dependence CGRP怎样受体拮抗剂(摩尔和萨尔瓦多,2012年;沃克et al ., 2015)。孵化HEK293细胞表达人类CGRP怎样受体ubrogepant封锁了α-CGRP-stimulated阵营回应,IC500.08纳米。浓度增加ubrogepant平行向右的变化引起的αcgrp怎样剂量反应曲线在营里功能试验和dose-ratio情节显示一条直线。此外,116年放映了目标表明,ubrogepant CGRP怎样受体高选择性,亲和力较弱的多巴胺转运体(K我4440海里)。这种多巴胺活动可能是药物无关紧要的血浆浓度预测是有效剂量。
恒河猴CIDV模型中,辣椒素激活草酸受体1,生产通过激活背根神经源性炎症和血管舒张反应和血管活性的介质的释放,这是主要由CGRP怎样驱动。这种反应可以被CGRP怎样受体拮抗剂,因此允许体内ubrogepant效力的评估与内生CGRP怎样发布时(Dux et al ., 2003;好时et al ., 2005)。基于抑制的PK / PD关系CIDV ubrogepant,估计指的是电子商务50和电子商务90年值分别为3.19和29海里。人口PK / PD CIDV建模在当下恒河体内研究表明,E马克斯为抑制CIDV ubrogepant为0.732,表明CGRP怎样是主要的,但不是唯一,贡献者CIDV (p物质在血管舒张和组胺可能也扮演了一定的角色),通常与其他CIDV研究结果一致CGRP怎样化合物(Vu et al ., 2017)。也观察到类似的结果人类研究使用CIDV模型和导致类似的估计意味着电子商务50和电子商务90年的值分别为2.56和23海里。综上所述,这些快速评估CGRP怎样受体在非人灵长类动物和人类对抗活动参与者表明预测PK-PD ubrogepant跨物种之间的关系。
在中枢神经系统渗透研究,CSF:等离子体比率为0.03。有限的渗透进入中枢神经系统表明ubrogepant不容易穿过血脑屏障,这是由目前的受体占用数据显示中央CGRP怎样受体入住率低(0% - -16%)等离子体水平的53 - 203海里。虽然确切的网站CGRP怎样受体拮抗剂的作用还不得而知,有限的ubrogepant渗透到中枢神经系统和概念是一致的,在神经与血管的头痛、敏化和活化trigeminovascular系统导致血管周的神经肽的释放,如CGRP怎样(戈德比et al ., 1988;何鸿燊et al ., 2010)。三叉神经节位于外的血脑屏障,因此很容易影响CGRP-focused治疗(Eftekhari et al ., 2015)。ubrogepant的有限的中枢神经系统的活动可能是有益的在避免潜在的副作用中央CGRP怎样对抗,和这个有限的中枢神经系统活动的潜在临床效益是未知的。
除了CGRP怎样的高亲和力受体,ubrogepant显示关联的艾米1受体,并在较小程度上,艾米3受体。100倍的区别在ubrogepant CGRP怎样受体和艾米之间的效力1受体可能代表的差异αCGRP怎样绑定两个受体,这表明ubrogepant可能有一个结合位点与CGRP怎样。CGRP-responsive艾米的识别1受体在三叉神经节神经元和CTR和RAMP1蛋白的表达在脊髓三叉复杂表明这些受体的作用的中央处理CGRP怎样信号(沃克et al ., 2015)。此外,动物基因敲除研究已经确定了pro-nociceptive角色艾米1(Gebre-Medhin et al ., 1998)。然而,艾米的角色1受体在偏头痛在很大程度上仍是个未知数。
抑制CGRP怎样受体已成为一个有前途的偏头痛的急性和预防性治疗的目标(Edvinsson 2018)。先前研究小分子CGRP怎样拮抗剂已经证明疗效治疗偏头痛;然而,临床研究涉及telcagepant和mk - 3207显示潜在的肝酶,担心药物引起的高程和临床发展的这些化合物是停止(休伊特et al ., 2011;何鸿燊et al ., 2014;哈格里夫斯和奥尔森,2019)。虽然这个肝毒性的确切机制尚不清楚,是假设部分归因于活性代谢产物的形成,而不是特定于CGRP怎样受体拮抗(哈格里夫斯和奥尔森,2019)。没有任何ubrogepant-associated肝毒性已被安全数据支持从最近的临床研究(戈德比et al ., 2019;哈钦森et al ., 2019)。
单克隆抗体CGRP怎样和CGRP怎样受体已经证明了在偏头痛患者疗效;然而,这些药物注射药物批准用于偏头痛的预防治疗(珀et al ., 2017;Ajovy(包插入)2018;Aimovig(包插入)2018;Emgality(包插入)2018;2018 a, b, c;Dodicket al。,2018年;Staufferet al。,2018年)。功效为预防治疗一般以月间隔,因此偏头痛发作的治疗方法仍需要快速救援(珀et al ., 2017;Ajovy(包插入)2018;Aimovig(包插入)2018;Emgality(包插入)2018;2018 a, b, c;Dodick et al ., 2018 b;Stauffer et al ., 2018)。Ubrogepant是口服CGRP怎样受体拮抗剂批准用于偏头痛的急性治疗能够提供自由从疼痛2小时(Dodick et al ., 2018;立顿et al ., 2018;沃斯et al ., 2016)。急性治疗偏头痛发作的主要管理和在一些病人可能补充预防治疗头痛频率等因素的基础上,急性治疗反应和migraine-related残疾(戈德比斯派格,2010)。此外,口服给药途径ubrogepant可能是首选的病人需要多个急性治疗偏头痛发作,而注射或输液给药途径为舒马曲坦和双氢麦角胺(O 'Quinn et al ., 1999,D.H.E. 45包插入2002)。Ubrogepant因此代表了一类新的药物治疗偏头痛发作的急性治疗。
总之,ubrogepant是一个强有力的,有选择性的,口头传递,人类CGRP怎样的竞争抑制剂小分子受体,显示了一个可预测的PK-PD关系和有限的渗透穿过血脑屏障在临床上有效的风险敞口。
确认
作者要感谢Jan胡昂,玛伦迪普雷(从中心临床药理学,鲁汶大学医院和药品药理科学KU鲁汶,比利时),汤姆Reynders (MSD欧洲公司,布鲁塞尔,比利时),和尤金•莱(Merck & Co。公司进军NJ)为人类capsaicin-induced真皮血管舒张试验模型设计和行为,和约翰广场(默克公司& Co .公司,进军NJ)为人类capsaicin-induced真皮血管舒张试验数据分析。写作和编辑援助被Peloton优势,提供给作者LLC新泽西,帕西帕尼由艾尔建公司资助。在这篇文章中表达的观点的作者。作者没有收到酬金/费用或其他形式的金融支持本文的相关发展。作者要感谢莫娜珀塞尔进行猴子宠物研究中,威廉姆斯Mangey分析支持,Aniket Joshi宠物数据分析,写作手稿和Abhijeet Jayate援助。
作者的贡献
参与研究设计:摩尔,贝尔,Burgey Li Hostetler,萨尔瓦多。
进行实验:摩尔Fraley,白色,里根,丹齐格,麦切纳。
了新试剂或分析工具:Hostetler。
执行数据分析:李。
或导致写作手稿中写道:摩尔Fraley,贝尔,Burgey,白色,李,里根,丹齐格,麦切纳,Hostetler,巴纳吉,萨尔瓦多。
脚注
- 收到了2019年8月12日。
- 接受2019年11月18日。
这项工作是由艾尔建公司,都柏林,爱尔兰。
这项工作曾提出以抽象的形式:摩尔E, Burgey CS, Fraley M,丹齐格,里根C,李CC,白色的RB, Banerjee P,萨尔瓦多C (2019)。表征ubrogepant:一个强大的和有选择性的人类降钙素相关基因肽受体的拮抗剂。首页。92年增刊15:p4.10 - 021。
披露:E.M.,M.E.F.,I.M.B.,C.S.B.,R.B.W.,C。-C.L., C.P.R., A.D., M.S.M., E.H., and C.S. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.,, (Kenilworth, NJ,) and own/hold stock/stock options of Merck & Co., Inc., (Kenilworth, NJ.) P.B. is an employee of Allergan plc (Madison, NJ) and owns stock in the company.
↵
 本文在补充材料jpet.aspetjournals.org。
本文在补充材料jpet.aspetjournals.org。

缩写
- 我
- adrenomedullin
- 艾米
- 糊精
- 营,环磷酸腺苷;CGRP怎样
- 降钙素相关基因肽
- CIDV
- capsaicin-induced真皮血管舒张
- CLR
- 降钙素受体受体
- 中枢神经系统
- 中枢神经系统
- 脑脊液
- 脑脊髓液
- CT
- 降钙素
- CTR
- 降钙素受体
- HEK293
- 人类胚胎kidney-derived
- hERG
- 人类ether-a-go-go-related基因
- PD
- 药效学
- 宠物
- 正电子发射断层扫描
- PK
- 药代动力学
- 斜坡
- 受体活性修饰蛋白
- 版权©2020年作者(年代)。
这是一个开放的分布式下文章CC归因4.0国际许可证。





{kind=link}
{kind=link}
{kind=link}
{kind=link}