条文本

文摘
肌萎缩性脊髓侧索硬化症(ALS)的发病通常被认为是开始与临床症状的识别。我们建议,与其他神经退化,致病机制最终ALS表型在生活中更早开始。动物模型的由基因决定的ALS展览病理异常长比临床赤字。公开临床ALS表型可能发展当安全利润超过随后的年的线粒体功能障碍,神经炎症或神经纤维网的兴奋和抑制不平衡环境。体细胞突变,表观基因组和外部环境影响可能引发代谢级联,在成年人的交互最终超过功能的阈值。很长一段时期发生前症状临床前期和后续识别构成挑战,因为它提供了一个机会,保护,甚至预防治疗拯救功能失调的神经元。我们建议,通过类比与其他神经退化和SOD1 ALS老鼠研究漏洞可能诱导产期。
- 肌萎缩性侧索硬化症
- 神经毒理学
- 神经
- 运动神经元疾病
- 分子生物学
来自Altmetric.com的统计
介绍
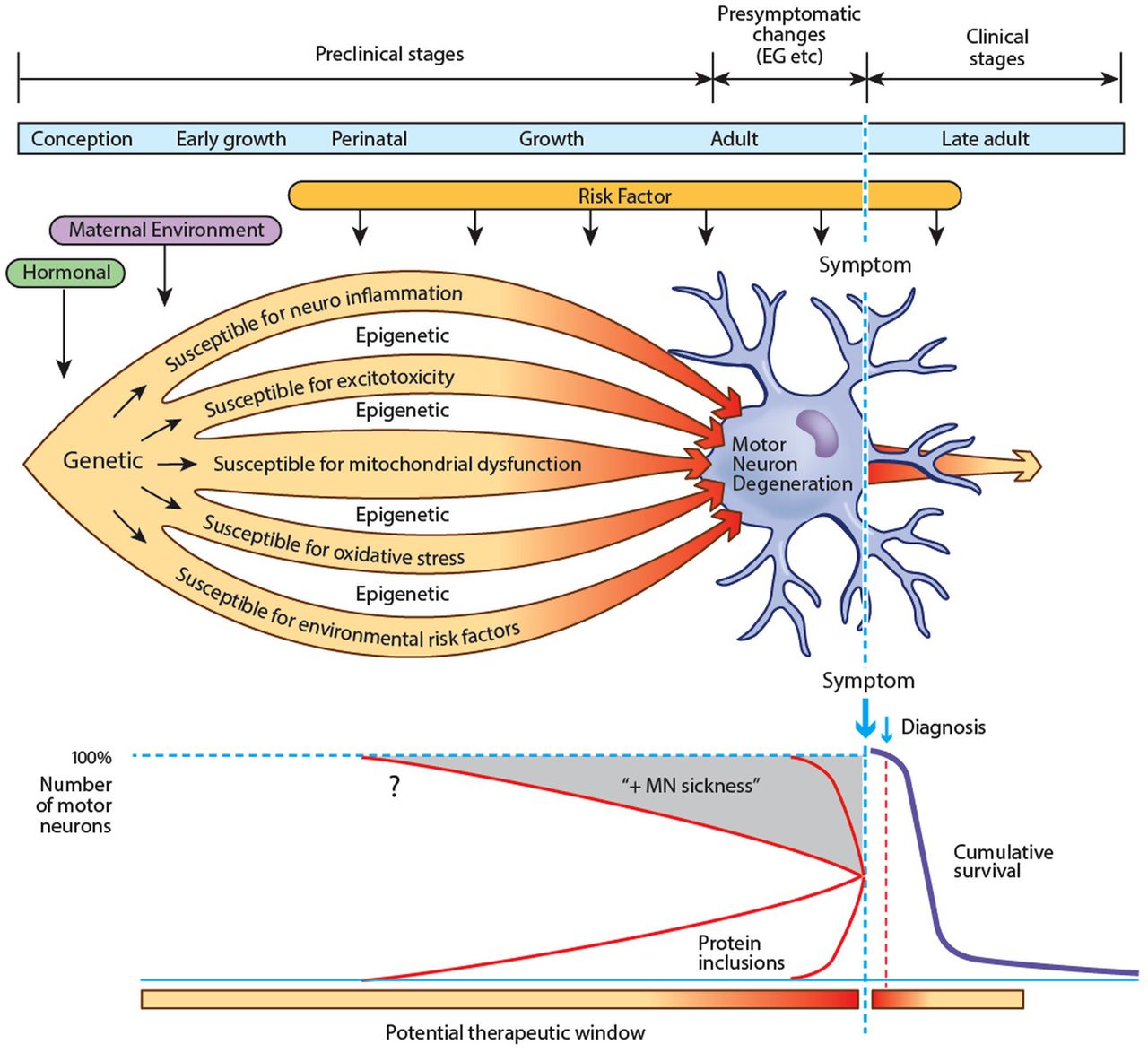
肌萎缩性脊髓侧索硬化症(ALS)似乎难以治疗的努力。这令人失望的治疗反应可能只是反映了表型表达引发更早,也许几十年的临床症状出现之前。1条款发生前症状临床前和经常交替使用,但是这里发生前症状的”指的是时期没有临床相关,而神经成像等调查,电生理或认知评估可能异常。“临床”指的是更长时期目前还没有确定的标记在零星的ALS疾病图1)。
{kind=link}
从生物学的角度,种子发展的肌萎缩性脊髓侧索硬化症(ALS)可能是播种后不久的观念。运动神经元和支持神经胶细胞易受许多潜在的侮辱,如神经炎症,会引起线粒体功能障碍,过度的氧化应激和环境危险因素。表观遗传的影响可能会进一步确定个体敏感性和易感性。环境风险因素一生中继续发挥他们的影响力。相结合,这些因素导致蛋白功能障碍和聚合。运动神经元和周围星形胶质细胞的新陈代谢强调,逐渐失去功能(MN“病”)。几年或几十年之后,胞质补偿机制开始失败,临床上可识别的阶段开始,电生理学和成像异常发生前症状成为检测在宏观层面上。最后,运动系统失败和ALS症状和不断进步。如果是这样,生物标志物可能变得明显在发生前症状临床前和阶段,从而使保护的未来发展或预防性治疗(例如,表观遗传的影响)。发生前症状的”一词指的是时期没有临床相关,而神经成像等调查,电生理或认知评估可能异常。 ‘Preclinical’ refers to the much longer period when presently there are no identified markers of disease in sporadic ALS.
症状出现在成人神经退化,包括肌萎缩性侧索硬化症,通常发生在中年后期的生活。在阿尔茨海默病(AD)和帕金森病(PD),病变临床疾病之前数年,如果不是数十年之久。在这两个广告2 - 9和PD,10 - 15之前有一段漫长的先兆的公开的功能开发。2 - 9非机动车在PD,有一段时间的前兆症状不典型的帕金森病中路易小体可能会发现在许多年前经典临床PD的发病。10 - 15甚至有人建议PD可能开始在围产期环境和遗传的影响可能降低多巴胺神经元的阈值,使正常功能继续前几十年的病理生理阈值超过临床表现的疾病。16
它曾被建议ALS可能长期临床前时期,17日至19日但是,一般来说,假设肌萎缩性侧索硬化症的临床发病是重合的,或开始后不久,该病发病病理过程的底层。证据,这涉及到一个短,期间发生前症状有减少运动单位的数量估计和肌电图(EMG)异常。20.,21同时,肌电图异常在临床上很常见强劲肌肉明显的疾病。22
然而,这些测试发现没有察觉的改变较低的运动神经元函数并不一定等同于前角细胞的正常功能;上运动神经元功能的异常显然已被证实之前在肌萎缩性侧索硬化症临床赤字。23更有可能的是,在细胞水平上生物分子功能障碍,目前无法检测到,可能不足以导致临床特征,但现在和建筑前几年或几十年临床疾病的发作。在SOD1肌萎缩性侧索硬化症小鼠模型,病理变化在出生后不久,很明显比第一个临床异常2 - 3个月。在人类基因联系ALS(歧视),disease-causative蛋白的表达,或其他代谢缺陷,即使在胚胎生命必须明显。同样,在零星的肌萎缩性侧索硬化症、生物异常反映了长期病态过程发展多年,甚至可能几十年,之前变得明显(见第一个症状图1)。在这里,我们将探索这种可能性与潜在的早期生理变化可能在中年预测ALS的可能性将开发到后期易感个体的生活。在这方面,遗传ALS是一个原型。
最早的异常肌萎缩性侧索硬化症转基因小鼠
啮齿动物模型的ALS不忠实地翻译成人类的疾病,尽管他们确实揭示临床pathobiological异常。24,25这些在理解相关类似人类肌萎缩性侧索硬化症早期异常,尤其是补偿机制,延缓临床表现。胚胎形态学和生理异常突变运动神经元已经显示包括兴奋过度。26Vinsant和他的同事们27,28分析了早期的变化SOD1G93A鼠标,包括超微结构检查神经肌肉系统的中心和外围组件和相关的这些变化与早期肌肉去神经运动功能障碍和运动神经元死亡。线粒体肿胀,有液泡的在脊髓和mega-mitochondria首次观察到产后7天。这些变化是在运动神经元树突最丰富,但在运动神经元soma还发现,神经肌肉突触前终端的连接和axo-somatic突触的突触前终端。积累的小空的空泡在脊髓运动神经元胞浆观察出生后14天。这些30多天了,后来细胞质变得充满了这些液泡。产后一天30,显著降低axo-somatic类型我在运动神经元的兴奋性突触和c端增加。没有变化II型的抑制性突触的数量和总数量的突触。临床上,步态改变和肌肉无力出生后30天开始,到60天,20%的运动神经元经历了变性。
神经发生前症状在hSOD1特定类型退化G93A老鼠是脊髓运动神经元和corticomotoneurons。29日中间神经元和non-neuronal元素,包括胶质细胞,也影响发生前症状的阶段变异啮齿动物。30.- - - - - -32转基因鼠脊髓运动神经元的电特性异常出生后不久,到三十五与突触变化有关,和改变神经回路和网络活动,其次是临床上明显的神经系统障碍。
胚胎和围产期的影响
很可能有一些零星的ALS的不明身份的风险基因。从单细胞受精卵发展成熟的生物带来大量的细胞分裂。因此,突变基因是频繁的在正常的人体发育进程中,尽管大多数不产生有害的影响。36适用于29日核基因遗传突变,目前已经确定与遗传有关肌萎缩性侧索硬化症,患病的风险最高,因为所有的细胞都携带着基因突变。37-42然而,自发与细胞分裂相关基因突变在胚胎发生和早期发展也可能disease-inducing。自发突变模糊遗传和零星的ALS的区别。43,44在零星的肌萎缩性侧索硬化症,自发地产生“风险突变”,在发展早期发生,只能携带一个稍低风险相对于遗传性肌萎缩性侧索硬化症。36自发突变产生后对神经系统发育较少,或者小的风险,这取决于早期或晚期的发展。突变基因如何转化为肌萎缩性侧索硬化症临床表型仍有待阐明,但很可能只有少数的众多基因的突变指导发展编程和网络形成和功能的整体负担将增加的风险在以后的生活中形成肌萎缩性侧索硬化症。45
神经网络发展从概念开始,并持续到青春期和成年早期。46然而,相关的产前和围产期时期最大的代谢活动。这是所需的神经发生,神经元增殖和神经分化和迁移。在人类,大多数编程神经元(凋亡)发生造成损失。增加的代谢需求增加氧化应激和必须由抗氧化剂生产和反击redox-sensing系统足以控制活性氧(ROS)生产,并移除受损的线粒体。47期间,自发突变可能导致微妙的异常在中枢神经系统(CNS)连接,连接和网络诱导脆弱性形成其“晚年”实现如此出色神经退化,包括肌萎缩性侧索硬化症。4后天,突触扩散的过程继续通过童年中期和紧随其后的是程序消除突触。实质性改进大脑的结构和功能发生在青春期,又一段潜在的疾病易感性。48
进化可能不会选择对早年生活的有害突变生效,只有在生殖时期。49虽然长寿过去生殖时期是重要的在我们的物种,“祖母效应”,某些遗传性状可能表现出拮抗基因多效性或表型改善生存在早些时候的生活,但随着年龄的增长而变得有害。50这是否适用于ALS还未确定。
临床前的遗传、表观遗传和环境风险
肌萎缩性侧索硬化症,像其他神经退化,是一个复杂的多因素疾病个体易感性的差异,年龄的发病和进展。遗传和环境因素影响的敏感性取决于多个gene-by-gene和gene-by-environment交互和表观遗传效应也开车表型的个性。51这些因素可能是疾病发生前症状解体的关键。
神经退行性疾病和孟德尔遗传疾病,包括家族性肌萎缩性侧索硬化症与遗传变异存在相关联的时间概念,即使他们不存在临床直到成年中期到后期。这意味着要么是这些基因不是“开启”,直到晚年,或者有几十年的进步细胞最终妥协灾难性的下降表现为临床上明显的肌萎缩性侧索硬化症。遗传研究表明,约60%的ALS的风险是由基因决定的,而剩下的40%环境决定的。52任何环境对ALS的贡献将不太可能单独行动;遗传和表观遗传组件都必须进行交互。
ALS的环境暴露作为一个风险因素,尽管显然薄弱的因素在疾病的因果关系,可能是累积随着时间的推移,超过那些genetic-environmental阈值,一些以后,患肌萎缩性侧索硬化症。神经退行性过程,此后,似乎是不可逆转的,追逐。高渗透环境暴露单基因导致几乎没有要求,但在一个复杂的oligogeneic或多基因疾病,如零星的肌萎缩性侧索硬化症、环境因素的影响较大。42
ALS发病率最高的是70和74岁之间;此后,发病率下降迅速。在这方面,ALS不同于AD和PD的发病率和患病率随着年龄的增加。的风险降低老年不是由于确定偏差,并可能部分反映了Gompertzian群的人对ALS是由环境和遗传风险因子之间的相互作用。53-55
环境暴露于毒素,吸烟,过度的体育活动,职业、饮食因素和免疫的变化都会增加发展中零星的ALS的风险。53这些因素可能多年来驱动表观遗传变化,从而引起疾病发生和发展。有一个重要的协会与吸烟;长期接触和当前吸烟ALS风险增加2到3倍。56,57暴露于污染物是一个机制,可能会触发并能长期维持神经炎症,但是否重复低接触可能与遗传和表观遗传成分的起始ALS尚未建立。42少关注针对更一般的环境因素,可能会引发运动神经元变性导致ALS的级联。58然而,神经损伤与氧化应激可能持续一生中积累的环境、职业、饮食和生活方式的风险敞口。59Neuroepidemiological ALS的风险因素的研究表明,接触必须发生在几年前疾病发作,43暗示一个环境触发可能多年活跃在临床疾病的发展。
表观遗传变化是发展和与年龄相关的生物学的基础。有前途的流行病学研究涉及表观遗传学疾病风险和进展,并建议表观遗传状态取决于环境风险以及遗传倾向。表观遗传学可能代表一个环境因素之间的关系和机制,修改所选基因的表达水平,在不改变DNA序列。这些机制包括DNA甲基化、组蛋白尾巴修改和染色质重塑,以及由小RNA分子机制。表观遗传修饰是很重要的,因为他们有类似的效果的致病突变,因为它们能够沉默,增加或减少选择基因的表达在一个给定的组织。60- - - - - -62年在开发过程中有一个关键的窗口中这些因素可以对神经元基因表达产生持久的影响。63年
表观遗传过程已确定在AD和PD。64 - 66在零星的肌萎缩性侧索硬化症,有人建议,表观遗传修饰可能改变pathogenesis-related基因的表达导致的发病和进展零星的ALS和ALS-dependent多个基因的甲基化之前与神经发育,分化和增殖。67年DNA甲基转移酶可调节运动皮层和脊髓运动神经元在零星的肌萎缩性侧索硬化症。68年因此,表观遗传缺陷体内平衡中枢神经系统,导致异常的基因表达,可能导致中枢神经系统功能障碍启动肌萎缩性侧索硬化症。61年
在ALS兴奋和抑制性神经传递
神经元可能呈现hyper-excitable当谷氨酸水平,增加或减少抑制,当γ-aminobutyric酸(GABA)活动,主要的抑制性神经递质,降低,或两者的结合。突变ALS啮齿动物,会引起胎儿所记载,它是公认的功能型人类零星的肌萎缩性侧索硬化症,26,69年虽然还未确定临床症状出现之前多久excitotoxic状态存在。70 - 72
一个hyperexcitable运动神经元会发射更多的响应给定的突触输入峰值,因此更多的钙离子流入细胞质中,最终导致神经细胞死亡。但是,与胚胎不成熟的运动神经元,内在兴奋过度从未成年豚鼠中演示。73年所以会导致ALS变性不是内在电特性的变化引起的豚鼠。然而,会引起的也可能是由运动神经元突触输入收到的变更。减少兴奋性输入的抑制性输入或增加将导致更高的发射率,从而增加营业额在细胞质中钙。73年
谷氨酸神经突的早期发展中是至关重要的产物,神经元迁移和大脑发展中经历了一段时间的过度刺激敏感性增加NMDA受体通道复合物。GABA出现ALS的发病机制的基础。74年在ALS,有广泛的小清蛋白的丧失和calbindin-D钙结合蛋白与GABA-ergic中间神经元。75年,76年减少抑制已被证明发生在使用核磁共振光谱学ALS运动皮层77年和经颅磁刺激已经确定减少短间隔皮层抑制突变与ALS的无症状携带者。77年,78年这些研究表明,超兴奋性先于ALS症状的发作。79年支持,正电子发射断层扫描使用配体flumazenil已经确定普遍减少脑-受体结合在肌萎缩性侧索硬化症。80年
GABA-mediated产后传播的主要作用是产生神经网络同步振荡。81年,82年脊髓抑制它目前尚未解决如果损失是导致肌萎缩性侧索硬化症或节段性神经破坏的结果。Renshaw细胞改变可能导致hyperexcitable最终状态和运动神经元变性。假定,这种兴奋过度造成的损失复发Renshaw细胞介导的抑制。83年另外,Renshaw细胞损失可能不是一个初始运动神经元兴奋过度和神经退行性变的因果关系,但运动神经元变性是次要的。83年,84年
神经炎症
越来越多的数据支持的假设不同的传染性病原体引起的损害可能会导致神经退化的因素。这可能在协同与其他危险因素,如老化,伴随的代谢疾病和主人的特定基因签名。85年不成熟和早产儿大脑可以暴露于病毒和细菌感染以及无菌侮辱发生在怀孕期间。这种炎症发作可能通常解决不损害中枢神经系统,然而,可能会增加对神经退行性疾病的脆弱性。86年,87年
在开发期间,小胶质细胞神经网络的形成做出贡献通过刺激vascularisation和协助修剪多余的神经元和突触,以及促进细胞分化。一生中,有一个平衡microglia-derived防护抗炎细胞因子,最大的早期发展和童年,与促炎细胞因子,积累与衰老和与慢性炎症状态有关。88年转向促炎细胞因子有助于增加易感性和神经退化。89年身体攻击在男孩的童年的预测减少了抗炎细胞因子在成年早期,90年提高ALS的有趣的猜测,男性的优势可能在一定程度上减少在生命的早期抗炎细胞因子有关。这可能配合发现ALS患者题为足趾率较低,符合产前循环的睾酮水平更高,可能是产前睾酮对运动神经元脆弱性的影响。91年
线粒体和mtDNA
线粒体是负责生成细胞能量,调节细胞内的钙含量,改变reduction-oxidation潜在的细胞和调节细胞死亡。92年,93年线粒体功能障碍,神经退行性疾病的早期事件,93年可以作为触发器或神经退化的宣传者。94年特别是,毒性从活性氧可以启动线粒体DNA损伤(mtDNA)导致呼吸链功能障碍,进而增加活性氧的生成,进一步促进细胞损伤,创建一个self-amplifying过程。95年
线粒体在细胞的数量变化与能源需求比例。神经元的高能源需求呈现它们的线粒体功能障碍。线粒体的数量、质量和本地化为神经元函数都是至关重要的。线粒体形态是由之间的平衡不断融合,线粒体在细胞可以互相支持,和裂变碎片的线粒体在细胞凋亡中起着重要的作用。线粒体动力学的变化被发现在许多神经退行性疾病,包括肌萎缩性侧索硬化症,假定,线粒体融合/裂变的失衡与疾病相关的线粒体功能障碍有关。92年,96年线粒体分裂被描述在转基因小鼠发生前症状97年和是人类ALS的早期特征。93年
mtDNA分子很小,对13种蛋白质编码,非常容易突变。98年因为线粒体的更新是一个非常活跃的过程,比核DNA mtDNA积累突变快得多,所以,致病性突变会影响许多mtDNA不同比例的分子从1%降至100%。99年突变可能是有利的在某些环境中但有害他人,所以形成的遗传基础的一部分潜在的复杂的疾病,如肌萎缩性侧索硬化症。One hundred.小的非编码小分子核糖核酸(microrna)已成为关键在调节基因表达及其在神经退化失调。miRNA-mediated监管活动的变更可能扰乱神经细胞所需的微妙的平衡发展和生存,从而导致疾病发生和发展。101年
结论:对研究和治疗的影响
我们假定ALS股票共性与其他神经退行性疾病中,有一个令人信服的证据表明发生前症状的临床症状出现之前是一个长时期。这样的一段时间可能持续几年或者几十年,与下游事件超过阈值仅供临床症状的出现变得明显年后pathobiological疾病过程开始。贝纳塔尔和Wuu,所强调的18确认这是可能对理解疾病生物学有着深远影响,揭示环境风险因素,发展有效的疗法,甚至疾病预防。
基因的研究晚发性神经退化,包括肌萎缩性侧索硬化症,收到太多的关注在过去的十年中,但表现的疾病表型之间的联系,改变了生物化学和细胞生物学检测到血液、脑脊液(CSF)或通过成像,以及microrna的表观遗传变化,仍然是模糊的。102年,103年我们建议许多不同的生物分子事件可能影响正常发展的这种疾病时才成为临床上明显的内在补偿机制分解,也许几十年后他们的发病。涉及的过程复杂,互动和进步。肌萎缩性侧索硬化症的临床综合征时神经元也可能astroglial成了明显的代谢被积累生物异常,尤其是能源动力学,直到到达一个“转折点”。施加的压力的困难新陈代谢蛋白质的废物,蛋白酶体和细胞质TDP-43积累所示,是一个底层的标志,但目前知之甚少异常。该病临床当细胞开始“落悬崖”成一个不可逆转的终端级联,导致细胞死亡。
因此,当前治疗未能有效地修改ALS在很大程度上可能反映了之间的长时间运行出现明显的病理过程和发病症状的疾病。因此就必须确定致病蛋白在临床前阶段的主要目标通过建立诊断工具来识别高危人群发生前症状患肌萎缩性侧索硬化症。104年此外,疾病发生前症状理解状态识别是至关重要的补偿机制,允许明显正常的脑功能,尽管持续的神经退化。漫长的时期发生前症状与受损细胞和神经网络功能障碍有关,可能出现在围产期,打开一个潜在的重要窗口可能允许救援功能失调的神经保护干预但尚未死亡的神经元。甚至有可能出现很多的代理之前进行,未能显示明显的肌萎缩性侧索硬化症受益,如果早期,可能神经保护属性。发育方面的背景下肌萎缩性侧索硬化症临床历史和量化中外部环境的影响特性,已经被证明是有用的在理解自闭症谱系障碍,可能会产生进一步的关键的见解,特别是关于引入潜在的神经保护治疗的最佳时间。105年
引用
脚注
贡献者每个作者的贡献同样的概念,编写和审查的手稿。
相互竞争的利益一个也没有。
出处和同行评议委托;外部同行评议。