

摘要
背景及目标重症肌无力(MG)是一种以神经肌肉连接处功能障碍为特征的自身免疫性疾病。治疗通常包括皮质类固醇(CSs)和IV免疫球蛋白(IVIG)。本研究旨在确定免疫球蛋白(人),10%辛酸/层析纯化(IGIV-C)是否可以促进CS依赖MG患者的CS剂量减少。
方法在这项随机双盲安慰剂对照试验中,cs依赖的MG患者(美国重症肌无力基金会II-Iva;AChR+)在2天内接受2g /kg IGIV-C负荷剂量(最大80 g/d)或在第0周(基线)接受安慰剂。维持剂量(1 g/kg IGIV-C或安慰剂)每3周给予一次,直到第36周。CS开始于第9周,持续至第36周,除非患者病情恶化(定量MG评分较基线≥4分)。加重患者的CS剂量增加(基于当前CS剂量)。如果恶化在6周内未能改善或需要第二次提高CS,则患者被撤院。主要疗效终点(第39周)为CS剂量减少≥50%。在整个研究和随访期间(第42周和第45周)评估次要终点和安全终点。研究结果和完整的方案可在clinicaltrials.gov / ct2 /显示/ NCT02473965。
结果主要终点(CS剂量减少≥50%)在IGIV-C治疗(60.0%的患者)和安慰剂(63.3%的患者)之间没有显着差异。次要终点无显著差异。安全性数据表明IGIV-C具有良好的耐受性。
讨论在这项研究中,IGIV-C在减少每日CS剂量方面并不比安慰剂更有效。这些结果表明IGIV-C和CS的作用不是协同的,可能在机制上是不同的。
试用注册信息该试验已在临床试验注册簿上注册。eu (EudraCT #: 2013-005099-17)和clinicaltrials.gov(标识符NCT02473965).
证据分类这项研究提供了II类证据,与安慰剂相比,成年MG患者输注IVIG不会增加患者实现皮质类固醇剂量减少≥50%的百分比。
术语表
- AE=
- 不良事件;
- CS=
- 皮质类固醇;
- IGIV-C=
- 免疫球蛋白(人),10%辛酸/层析纯化;
- 知识产权=
- 临床实验的产品;
- 丙种球蛋白=
- 静脉注射免疫球蛋白;
- LOCF=
- 最后的观察结果结转;
- MC=
- 肌无力的危机;
- 毫克=
- 重症肌无力;
- MG-ADL=
- mg -日常生活活动;
- MG-QOL=
- mg -生活质量仪器;
- 评分=
- 定量毫克;
- SAE=
- 严重的AE;
- TEAE=
- 治疗诱发的AE;
- WOCF=
- 最糟糕的观察结果结转
重症肌无力(MG)是由自身免疫介导的神经肌肉连接处功能障碍引起的。这种功能障碍通过突触后蛋白的自身抗体表现出来,通常是乙酰胆碱受体(85%-90%),较少的是脂蛋白相关蛋白4和肌肉特异性激酶。1,2
对胆碱酯酶抑制剂无反应的MG,主要治疗是免疫抑制。首选药物通常是皮质类固醇(CSs)。严重MG或加重的治疗通常包括血浆交换或IV免疫球蛋白(IVIG)。3.,-,5血浆交换被证明可以改善MG患者的肌肉力量。6,7IVIG的治疗效果与PE相当,但不良反应较少。8,-,10IVIG在MG加重期的临床益处被纳入许多神经学会的临床指南,并被考虑作为急性MG治疗的核心组成部分。11,12
尽管CS是一线免疫抑制治疗,长期使用CS与潜在的严重副作用相关。由于这一缺点,逐渐减少CS到最小有效剂量是MG管理的目标。13然而,在不恶化潜在MG的情况下减少剂量往往是具有挑战性的。此外,还没有标准的缩减指导方针。在这项研究中,在CS依赖的MG患者中测试了免疫球蛋白(人),10%辛酸/色谱纯化(IGIV-C),以确定与安慰剂相比,IGIV-C给药是否可以增加CS剂量减少≥50%的患者的百分比。
方法
研究设计
这项2期研究是一项多中心随机双盲安慰剂对照试验,在8个国家的24个中心进行,这些中心筛查和/或招募了患有MG的cs依赖患者。这些地点分布在加拿大、捷克共和国、爱沙尼亚、德国、匈牙利、立陶宛、波兰和美国。主要目的是评估IGIV-C与安慰剂(0.9%氯化钠无菌注射液,美国药典[USP]或等效物)相比,在减少cs依赖MG患者的CSs维持剂量方面的疗效。IGIV-C作为初始负荷剂量(2 g/kg)5,10,14,15随后每3周给予12次维持剂量(1g /kg)15,16(直到第36周)。主要终点是第39周CS剂量(强的松当量)较基线/第0周减少50%或以上的患者百分比。17该研究分为4个阶段:(1)筛选,(2)试验性产品(IP)磨合维护期,(3)CS逐渐减少IP维护阶段,(4)安全/随访阶段。患者按1:1随机分为IGIV-C组和安慰剂组。随机分层按CS基线剂量(15-40 mg/d强的松当量或41-60 mg/d强的松当量)。
治疗
随机接受IGIV-C治疗的患者在基线随访(第0周)时接受负荷剂量(2 g/kg) (图1).装载剂量分为2天,由于体重较高(限制80克/天)或为了增加耐受性,可允许最多4天。维持剂量为1 g/kg,超过1天,每3周给予一次,直到第36周。允许较长的给药期(2天)以允许较高的剂量(最大剂量80 g/d)或耐受性调节。随机分配到安慰剂组的患者接受等量的生理盐水(0.9%氯化钠,USP)。IGIV-C和安慰剂在加载剂量和维持剂量时双盲。
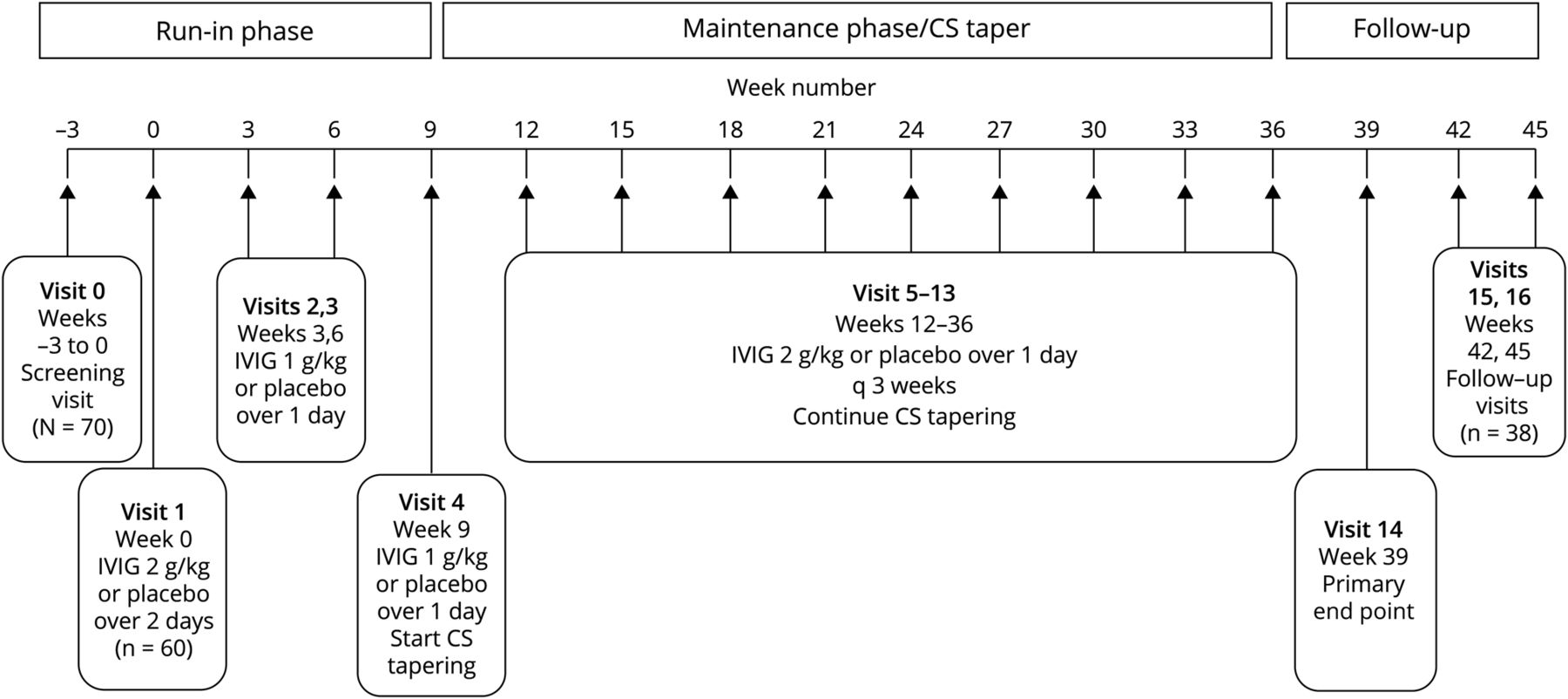
在注射3次IGIV-C或安慰剂(第9周)后开始逐渐减少CS剂量。如果患者的CS剂量为40 mg强的松(或等效)/天,每次就诊时(每3周)剂量减少10 mg(或等效)/天。如果患者的CS剂量≤40 mg强的松当量/天,剂量每3周减少5 mg当量/天。每隔一天服用CS的患者剂量逐渐减少,例如,如果>80 mg/每隔一天,每3周减少20 mg。最终CS锥度为0 mg强的松当量/天,由研究人员自行决定。研究人员试图持续维持非cs MG药物,除非患者经历了治疗的不良反应或MG的恶化。加重定义为患者定量MG (QMG)评分较基线增加≥4分。17
如果在CS逐渐减少阶段发生MG恶化,则患者的CS剂量增加20mg(如果当前剂量≥15mg,则相当于强的松),如果< 15mg,则增加15mg。在每隔一天给药CS的情况下,患者的CS剂量相应增加,例如,如果当前剂量≥30 mg,则增加40 mg。增加的剂量维持了6周(接下来连续2次就诊)。如果患者的MG稳定,定义为患者的QMG评分相对于基线(第0周)增加≤3分,则允许患者继续研究。如果MG恶化在剂量增加后的6周内没有改善,则患者退出研究。
如果增加CS剂量成功地改善了QMG评分的恶化(较基线增加≤3分),且患者的临床症状恢复到基线,则进行第二次CS逐渐减少尝试。在第二次尝试时,CS剂量没有低于先前观察到症状恶化的剂量。任何症状需要第二次增加剂量的患者都退出了研究。
研究患者的选择
年龄18-85岁,抗achr抗体阳性,确诊为全身性MG(美国重症肌无力基金会[MGFA] Class II, III, IV, V)的男性和女性患者符合筛查条件。18筛查时,要求潜在参与者的MG症状被CS控制,并有MGFA II-IVa史(MGFA IVb级和V级;仅排除眼部MG)。筛查前需要至少3个月的全身CS,稳定的CS(强的松当量)剂量≥15和≤60mg /d,持续1个月。对于每隔一天给药的潜在参与者,他们的剂量需要达到每日剂量标准的一半。在本研究中,这些标准定义了激素依赖性MG。研究者认为,逐渐减少患者的CS剂量在临床上是合适的,并且至少有过1次减少CS剂量的尝试。需要书面知情同意。
如果患者在筛查前6个月有任何非CS伴随免疫抑制治疗的变化,或在筛查前一个月有任何CS剂量或乙酰胆碱酯酶抑制剂剂量的变化,则排除在研究之外。筛查和基线(第0周)来访之间QMG评分的3点变化(增加或减少)为不合格。排除筛查前一个月有肌无力危象(MC)发作和任何MC史或因与CS减弱相关的MG加重住院治疗。其他排除的因素包括:过去5年恶性肿瘤、可能需要手术的胸腺瘤、过去6个月的胸腺切除术、心血管疾病史、肾损害、肝酶升高或贫血。
过去12个月内接受过免疫调节单克隆抗体治疗、过去3个月内进行过血浆置换或目前正在接受抗凝治疗者不符合入选条件。排除有MG对IVIG无反应史、筛查前3个月接受过免疫球蛋白治疗、不耐受或对IVIG过敏、对IVIG有血栓性反应或已知的高粘度或高凝状态。已知IgA缺乏症和抗IgA抗体的患者不符合研究条件。
临床实验的产品
本试验中使用的IGIV-C产品为Gamunex-C(免疫球蛋白注射液(人)10%辛酸/色谱纯化,Grifols Therapeutics, LLC, Research Triangle Park, NC)。15在本研究中,生理盐水(0.9%氯化钠无菌注射液,USP)或同等物质作为安慰剂。输注由未失明的药剂师或指定人员配制,因此安慰剂输注与IGIV-C输注难以区分。
研究终点
本研究的主要疗效终点是第39周CS剂量较基线(第0周)减少50%或以上的患者百分比。从基线(第0周)到第39周测量的次要疗效终点是CS每日剂量减少的百分比和MG首次恶化的时间(如上定义)。
探索性端点与CS治疗包括以下:实现减少≥75%的患者百分比CS剂量在39周,实现CS剂量的患者百分比≤7.5毫克(强的松当量)在39周,患者的百分比CS-free 39周,空腹血清葡萄糖的变化在39周与基线,空腹血糖的患者百分比≤125 mg / dL在39周与基线,和糖化血红蛋白的变化在39周与基线相比(周0)。
MG相关的探索终点如下:百分比的MG需要住院治疗的病人经历MC或恶化通过39周和39周星期45,MG恶化的事件数量从基线(周0)到39周,变化在15个MG-Quality生命的仪器(MG-QOL 15)在39周,42岁和45与基线(周0)相比,变化MG-Activities的日常生活(MG-ADL)得分在39周,42岁,从基线(周0)和45,从基线(周0)的活动(绑定、阻塞和调节)抗乙酰胆碱受体抗体在第39周(科文斯中心实验室,印第安纳波利斯,IN)。此外,在第9周、第24周和第39周测量血清IgG水平较基线(第0周)的变化。
CS锥度的指南是QMG评分。17QMG评分3分的改善反映了临床显著改善。19服用胆碱酯酶抑制剂的研究患者被指示在QMG测试前12小时内不要服用这些药物。MG-QOL 15是由患者评估的活动能力、症状、总体满意度和情绪健康的衡量标准。20.,-,22MG- adl评分旨在评估MG对日常活动的影响。MG-ADL改善2点被指定为临床显著性。23MG复合量表已被MGFA推荐作为全身性MG患者的定量测量。24,25MG复合的3点改善与临床改善和患者有意义的改善相关。26
安全评估
安全性评估包括报告所有不良事件(AEs)、严重不良事件(AEs)和因不良事件而中止试验。同时监测患者的血栓栓塞事件和溶血情况。在筛查时评估血栓栓塞风险,基线(第0周;输注前),完成第一次负荷剂量输注后,完成最后一次负荷剂量输注后,以及维持输注完成后的第3、6和24周(溶血评估在这些时间加上输注后1周)。
统计分析
改良意向治疗人群为疗效分析的主要人群,患者根据治疗方式进行分类。对改良意向治疗人群也进行了初步和二次分析。根据就诊时的处方剂量和考虑就诊期间剂量变化的时间间隔,计算每位患者的平均每日CS剂量。非分层治疗比较采用Fisher精确检验。采用Cochran-Mantel-Haenszel试验对CS基线剂量(15-40 vs 41-60 mg/d)进行分层治疗比较。采用协方差分析对次要疗效终点进行分析。退出研究的患者的缺失数据采用最后观察结转(LOCF)方法处理。
对于探索性终点,使用Fisher精确检验进行治疗比较,由于细胞小,没有调整分层基线强的松当量剂量。对于早期停止研究并出现MG相关不良结局的患者,使用最坏观察结转(WOCF)方法计算第39周时缺失的CS剂量。对于由于其他原因在第39周没有CS剂量的参与者,使用LOCF来计算第39周缺失的CS剂量。对来自安全人群的数据进行安全性分析,并进行描述性分析。
数据可用性
支持本研究结果的数据可根据合理要求从通讯作者处获得。研究结果和完整的方案可在clinicaltrials.gov / ct2 /显示/ NCT02473965。
标准方案批准、注册和患者同意
研究方案由所有参与机构的伦理委员会、机构审查委员会或研究伦理委员会批准(补充材料中有完整列表),并由所有参与国家的监管机构批准。所有参与者提供书面知情同意书。该研究是根据适当的当地法律法规、良好临床实践国际标准和赫尔辛基宣言进行的。该试验已在临床试验注册簿上注册。eu (EudraCT #: 2013-005099-17)和clinicaltrials.gov(标识符NCT02473965).
结果
治疗组基线特征
耐心的性格
这项研究筛选了24个地点的70名患者,其中60名是随机分组(意向治疗人群)(图2).所有60例患者被纳入改良意向治疗人群进行疗效分析和安全人群进行声发射分析。38例患者(63.3%)完成了所有研究访视。两个治疗组的完成数量相似:18例(60.0%)IGIV-C和20例(66.7%)安慰剂。
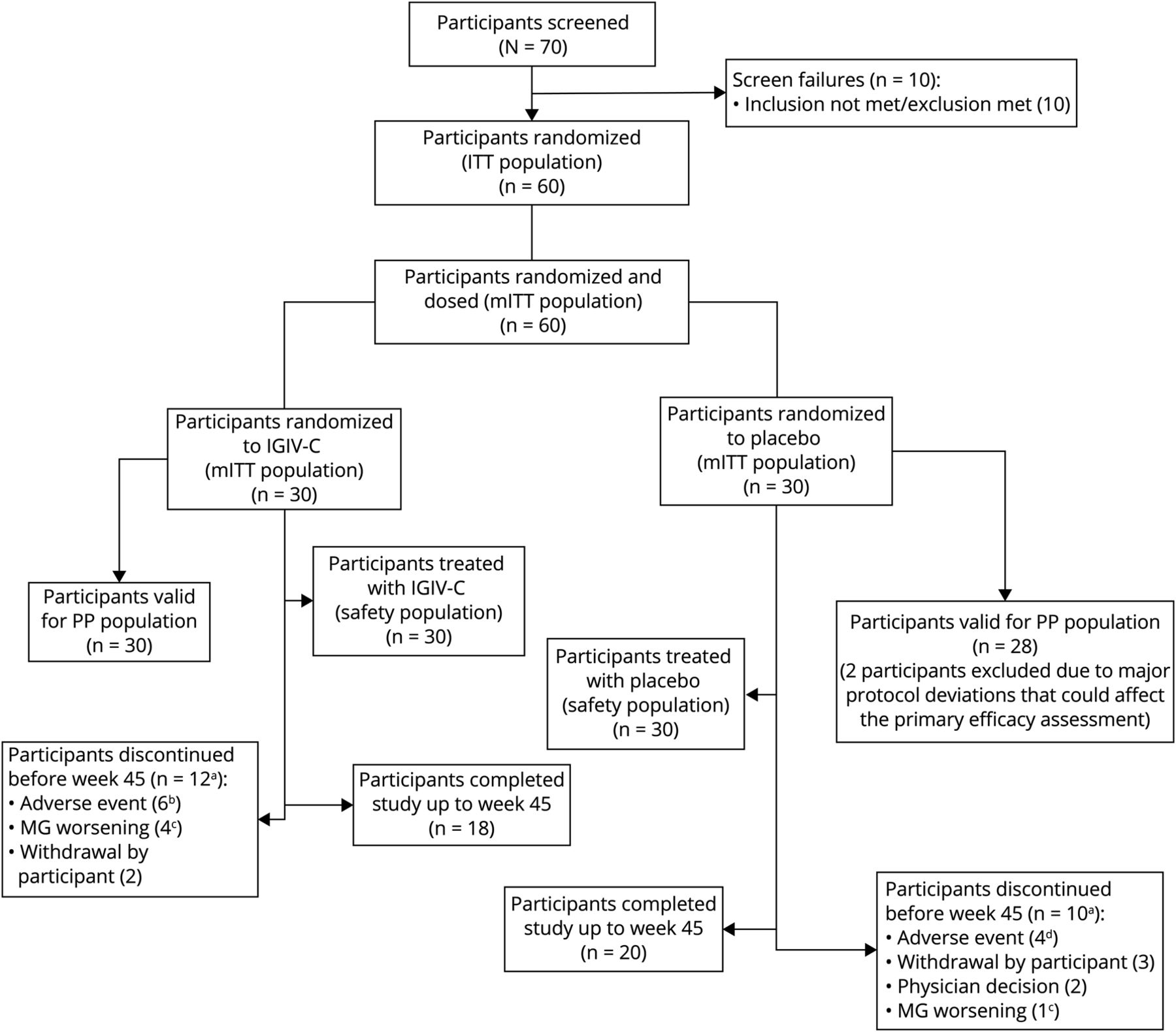
一个除3名参与者在第9周之前退出(1名参与者使用IV免疫球蛋白[IGIV-C], 2名参与者使用安慰剂)外,所有停用都有效地促进了皮质类固醇(CS)疗效的逐渐减弱终点,因为根据方案,CS的逐渐减弱直到第9周才开始。b不良事件包括重症肌无力加重(MG) (n = 4)、溶血(n = 1)、头晕(n = 1)。c该图中的MG恶化指的是由于CS减弱失败导致的协议强制停药:CS无反应或第二次发作指的是MG恶化。d不良事件包括镁相关表现(n = 3)和败血症(n = 1)。
在过早停用IGIV-C组的12例患者中,6例是由于ae, 4例是由于MG恶化,2例撤回同意(表3).在安慰剂组的10例过早停药中,4例是由于ae, 1例是由于MG恶化,3例是由于研究者决定(非ae)而撤回同意。MG恶化是指由于CS锥度失效而导致的方案定向停药(见治疗部分)。
疗效终点
主要疗效终点为CS剂量降低50%的患者百分比(第39周vs基线[0周])。在这一主要终点上,治疗组间无显著差异(p= 1.00)。在IGIV-C治疗组,60.0%的患者达到了CS剂量减少50%,而安慰剂组达到了63.3%的水平(图3).
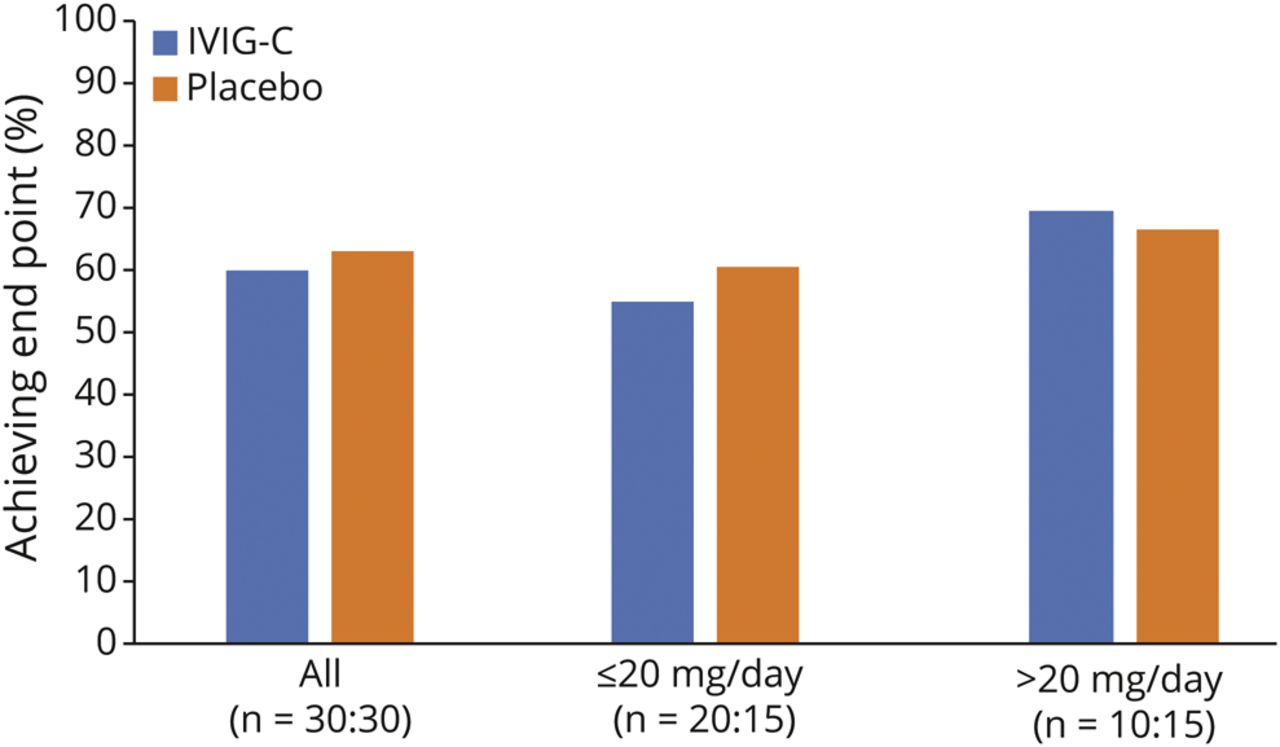
根据输入CS剂量是否处于或低于中位数(n = 20 IGIV-C;n = 15安慰剂)或高于中位基线剂量(20mg强的松当量)(n = 10 IGIV-C;n = 15安慰剂)。治疗组之间总体上没有显著差异。亚组表明,在两组中,如果进入高CS剂量分位数,则达到主要终点的百分比更高。IGIV-C =免疫球蛋白(人),10%辛酸/层析纯化;IVIG =静脉免疫球蛋白。
对主要终点的分析也对基线CS剂量分层的患者进行。然而,由于高CS剂量层(每天41-60 mg强的松当量)的患者数量非常少,低于预期(n = 1 IGIV-C;N = 2安慰剂),无法进行有效的统计分析。因此,在研究开始时规定的CS中位基线剂量的基础上进行了额外的分析。
患者按低于、等于或高于基线中位CS剂量(20mg /d强的松或等效剂量)的每日CS剂量进行分组。对于两个治疗组,高CS剂量(>20 mg/d)的患者在39周时比低CS剂量(≤20 mg/d)的患者更有可能实现50%的CS剂量减少。这一发现见于两组:IGIV-C: 70.0% vs 55.0%,安慰剂:66.7% vs 60.0% (图3),两组间均无显著差异(p= 1.00)。
本研究分析的次要终点是CS剂量减少百分比和MG症状首次恶化的时间。治疗组间CS剂量减少百分比无统计学差异(IGIV-C组减少52.04%±44.49%[平均±SD];安慰剂组降低54.69±51.36%)或首次恶化时间(第25百分位首次恶化时间[≥+4分QMG评分]33.10周IGIV-C;30.10周安慰剂)。
在研究期间,使用Kaplan-Meier方法计算MG随时间恶化的概率(图4).该分析显示治疗组间MG恶化的概率无差异(p= 0.744)。
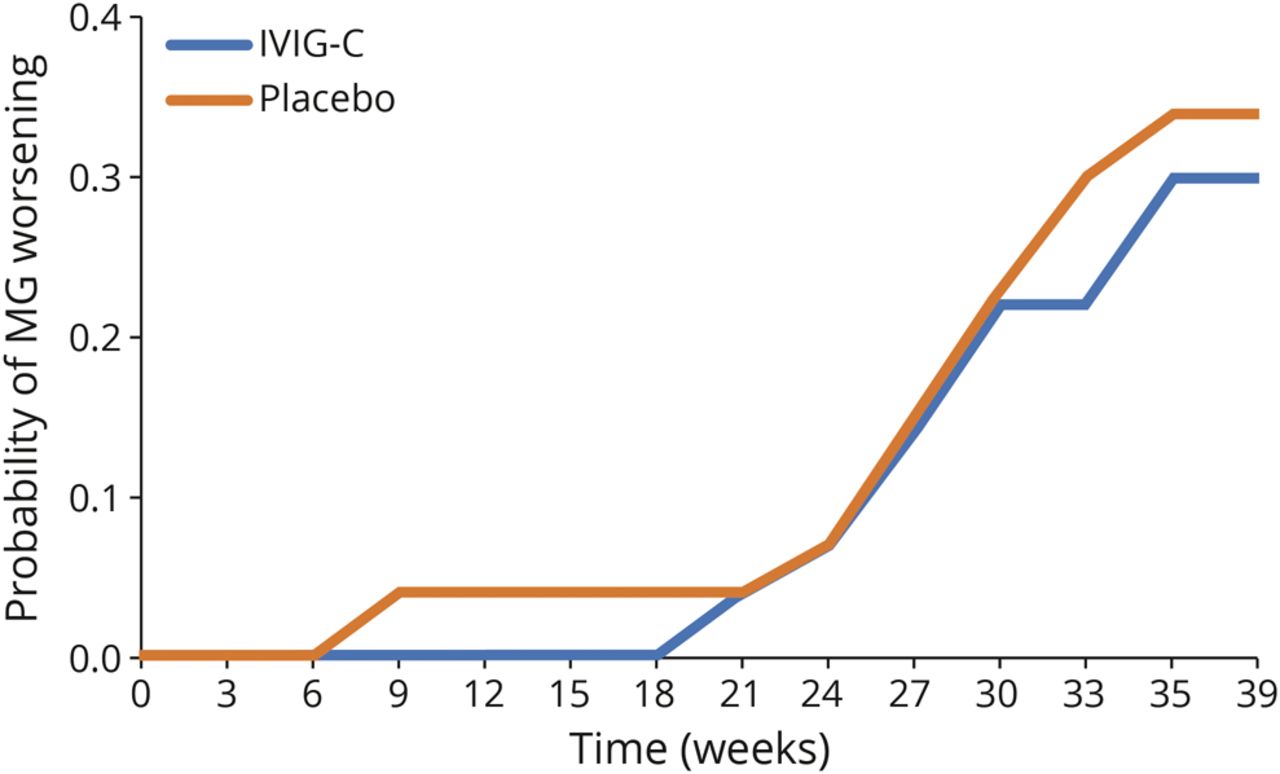
治疗组间无显著差异(p= 0.744)基于log-rank检验。MG恶化定义为定量MG (QMG)评分≥4分增加。
除IgG低谷水平外,两组探索性疗效终点无显著差异。IGIV-C治疗组的IgG谷水平明显高于安慰剂组。除第9周前3例外,所有停药均有效促进疗效CS逐渐减弱终点(1例有效;2安慰剂)。
安全终点
安全性数据显示IGIV-C治疗耐受性良好。两个治疗组的平均剂量、平均暴露时间和平均输注天数相似。
IGIV-C组90%(90.0%)的患者经历了至少1次治疗引发的AE (TEAE),与安慰剂组(93.3%)相似。IGIV-C治疗组最常见的teae(>15%)为头痛、MG加重、上呼吸道感染和恶心。在安慰剂组,最常见的teae是关节痛、背痛和鼻咽炎。
两组的teae大多为轻度或中度。严重teae是罕见的:IGIV-C: 4.5%,安慰剂:13.4%。
IGIV-C组4/30(13.3%)患者报告了sae,安慰剂组6/30(20.0%)患者报告了sae。IGIV-C组1例死亡,安慰剂组2例死亡。每臂均有1例死亡与MG相关。死亡原因包括MG急性加重、败血症、MG危象背景下的心脏骤停、葡萄球菌肺炎和急性呼吸衰竭。7例患者报告了MG加重或MG危象的SAEs: IGIV-C治疗组4例(13.3%),安慰剂组3例(10.0%)。
在IGIV-C治疗组中,有MG加重和/或MG危机的SAEs的4名参与者中,所有4人都完全逐渐脱离CS(无CS),在急剧恶化之前,MG在CS中保持稳定。在这4名参与者中,1名参与者在增加CS剂量和使用商业免疫球蛋白后死亡,1名参与者对8次血浆交换无反应,因长时间插管导致永久性残疾(气管狭窄)。
安慰剂治疗组共有3名MG加重/MG危在化的SAEs参与者,其中1名参与者在MG加重/危在化时完全逐渐减少到0 MG CS。虽然住院并接受了IVIG治疗,但CS没有重新引入,尽管进行了干预,他还是死亡了。另外2名MG加重/危像需要住院的SAEs安慰剂参与者逐渐减少到最低CS剂量,每天4 MG甲基强的松或10 MG强的松。
IGIV-C治疗组的30名参与者中有7人(23.3%)和安慰剂治疗组的4/30人(13.3%)有导致停药的不良事件。导致停药的不良事件最常见的是MG恶化、MG加重和MG危象。其他导致戒断的不良事件包括溶血、头晕、败血症和心脏骤停。
证据分类
这项研究提供了II类证据,与安慰剂相比,成年MG患者输注IVIG不会增加患者实现皮质类固醇剂量减少≥50%的百分比。
讨论
本研究的主要目的是确定IGIV-C是否可以促进MG患者CS依赖的CS剂量逐渐减少。与基线相比,IGIV-C治疗和安慰剂在第39周CS剂量减少≥50%的患者数量的主要终点上没有发现显著差异。值得注意的是,这一结果可能受到其中一项选择标准的影响:符合条件的患者必须至少完成了一次减少CS的尝试。这确保了患者使用尽可能低的CS剂量,并且在这些患者中CS剂量有可能减少。
次要终点分析显示,IGIV-C治疗对CS剂量或MG恶化首次发作的时间变化百分比没有显著影响。同样,探索性疗效测量在治疗组之间没有差异。总的来说,在这项为期36周的治疗试验中,IGIV-C治疗在促进MG患者CS剂量减少方面没有观察到优于安慰剂。
关于患者性格的研究设计的一个关键前瞻性因素是停药患者完全有助于CS剂量减少的疗效终点。根据方案,如果患者经历MG恶化(QMG增加≥4点),且对CS剂量增加无反应,或在第二次CS逐渐减少时再次恶化,则需要停止研究。CS锥度的预定义截断确保了在功效分析中充分反映了锥度失效,从而最大限度地减少了过早中断的影响。此外,在疗效分析中,使用了LOCF,对于所有与cs相关的终点,如果由于治疗引起的MG SAEs而停用,则使用WOCF。事实上,除3例在第9周之前停用(1例IGIV-C和2例安慰剂)外,所有停用患者都有效地促进了CS逐渐减弱疗效终点的实现。因此,过早停用不影响疗效结果的稳健性。
CSs是治疗对乙酰胆碱酯酶抑制剂不完全反应的MG的重要工具。一项回顾性分析显示,74%的MG患者对CS治疗有反应。16CSs治疗MG的疗效可能是由于其免疫抑制作用。27然而,长期使用CSs可能与许多严重的不良反应有关,包括体重增加、库欣综合征、糖耐量受损、血脂异常、高血压、骨质疏松症,以及极少数情况下的股骨头缺血性坏死。13,28可通过减少剂量来降低这些AEs的影响。13因此,CS治疗MG的目标是最小有效剂量。
IVIG也是MG的有效治疗方法3.,4在某些情况下,具有抗炎和免疫抑制作用。29确切的机制尚不清楚,但可能包括抑制树突细胞成熟,调节促炎细胞因子的产生,30.补体通路激活减少,31以及对巨噬细胞Fc受体的阻断。32这些机制使得IVIG在其他自身免疫性神经肌肉疾病的治疗中有用,例如,慢性炎症性脱髓鞘多发性神经病和多灶性运动神经病。
与其他免疫抑制剂的结果相似,33,-,35目前的研究表明,IVIG在降低CS剂量方面并不优于安慰剂。这些结果表明,单纯的免疫调节不足以促进剂量的减少。IVIG的抗炎作用也不允许剂量减少超过安慰剂。这些结果表明,CS对MG的作用在机制上是不同的,不能被IVIG的免疫调节特性所补偿。
本研究的持续时间(36周治疗,39周主要终点评估,45周随访)旨在评估CS剂量减少50%是否能在合理的时间框架内实现切实而有意义的获益。研究的CS逐渐减少部分的持续时间(从第9周开始到第36周结束)为27周或大约7个月。因此,本研究的主要疗效测试是稳定施用IGIV-C是否能够提供所需的治疗支持,使CS剂量在大约7个月内减少50%,这是评估将IVIG添加到现有MG方案的临床相关效果的实际时间周期。
IGIV-C没有产生明显优于安慰剂的益处的一个可能原因可能是安慰剂组本身在63%的时间内实现了≥50%的剂量减少。安慰剂组的这种改善可能很难证明IVIG的额外改善,而在其他MG试验中已经看到了这种改善。5,12,36这可能反映了相对最近的剂量变化对CS的额外免疫抑制作用,因为纳入试验的标准要求在入组前至少3个月的CS剂量,但只需1个月的稳定CS剂量。
在回顾性研究中,较长的随访时间有时是可行的,尽管这些研究缺乏同期对照,而且在患者管理和评估期间也可能有显著的差异。例如,最近的一项不受控的回顾性研究37显示长期IG给药(皮下或IV)显著降低CS剂量(<50%)。患者治疗15-78个月。这项研究发现,其他免疫抑制剂也可以减少CS的剂量。
本研究中观察到的teae与MG中IVIG和CS的其他研究相似。33,-,35,38,39IVIG在MG患者中仍然是安全的,可能与CS有独立的作用机制,而不是协同作用或相加作用。与安慰剂相比,IGIV-C在CS逐渐减少期间并未降低MG加重/恶化的发生率。两个治疗组中MG相关的sae在CS逐渐减少过程中强调,CS逐渐减少应缓慢进行,并仔细监测患者MG加重情况。
总之,本研究的数据表明,IGIV-C治疗在减少每日CS剂量方面没有优于安慰剂。然而,在两个治疗组中,较高的CS基线剂量(≥20 mg/d强的松当量)的患者比较低的基线剂量(<20 mg/d强的松当量)的患者更有可能实现50%的剂量减少。在较高CS剂量亚组中50%的减少基准可能允许残留的有益CS效应,因此更容易实现。
研究资金
这项研究是由Grifols资助的。
信息披露
V. Bril, A. Szczudlik, A. Vaitkus, C. Rozsa, A. Kostera-Pruszczyk, P. Hon, J. Bednarik, M. Tyblova, W. Koehler和T. Tomsoo报告没有与手稿相关的披露。R. J. Nowak获得了来自NIH, Genentech, Inc, Alexion Pharmaceuticals, Inc, argenx, Annexon Biosciences, Inc, Ra Pharmaceuticals, Inc(现为UCB s.a.),美国重症肌虚基金会(MGFA), Momenta Pharmaceuticals, Inc, immunnovant, Inc, Grifols, s.a.和Viela Bio, Inc (Horizon Therapeutics plc的一部分)的研究支持。他曾担任Alexion Pharmaceuticals, Inc, argenx, Cabaletta Bio, Inc, CSL-Behring, Grifols, s.a., Ra Pharmaceuticals, Inc(现为UCB s.a.), immunnovant, Inc, Momenta Pharmaceuticals, Inc和Viela Bio, Inc (Horizon Therapeutics plc的一部分)的顾问和顾问。T. Mozaffar, M. Friemer, M. w . Nicolle和T. Magnus报告没有披露与手稿相关的信息。M. Pulley曾接受以下区域咨询委员会和医疗咨询委员会(Alexion)的酬金:Gamunex(过去5年没有)、argenx、Amylyx、Alexion、BioProducts Laboratory、UCB/Ra Pharma、immunant、MT Pharma America和CSL/Behring。M.里夫纳报告说,没有披露与手稿有关的信息。M. Dimachkie目前或最近担任Amazentis、argenx、Catalyst、Cello、Covance/Labcorp、CSL-Behring、EcoR1、Janssen、Kezar、MedLink、Momenta、NuFactor、Octapharma、RaPharma/UCB、Roivant Sciences Inc、RMS Medical、赛诺菲健酶、Shire Takeda、Scholar Rock、Spark Therapeutics、Third Rock UCB Biopharma和UpToDate的顾问。他获得了来自Alexion, Alnylam Pharmaceuticals, Amicus, BioMarin, Bristol-Myers Squibb, Catalyst, Corbus, CSL-Behring, FDA/OOPD, GlaxoSmithKline, Genentech, Grifols, Kezar, Mitsubishi Tanabe Pharma, MDA, NIH, Novartis, Octapharma, orphhazyme, RaPharma /UCB, Sanofi Genzyme, Sarepta Therapeutics, Shire Takeda, Spark Therapeutics,肌炎协会,UCB Biopharma/RaPharma, ViroMed/Helixmith和TMA的研究资助或合同或教育资助。J. Distad和R.M. Pascuzzi报告没有披露与手稿相关的信息。D. Babiar, J. Lin, M. Querolt Coll, R. Griffin和E. Mondou是Grifols的员工,Grifols为本研究提供了资金支持,并且是10%人类免疫球蛋白,辛酸/色谱纯化(Gamunex-C)的制造商。 Go to首页Neurology.org/N全面披露。
鸣谢
Michael K. James博士和Jordi Bozzo博士在写作和编辑方面的帮助得到了认可。作者对参与该研究的患者表示感谢,以推进科学和医学,并对进行该研究的研究现场人员表示感谢。此外,作者还感谢了Grifols内部团队成员和合作进行这项研究的供应商。作者特别感谢陈俊良(Grifols)。
附录的作者

脚注
去首页Neurology.org/N全面披露。作者认为相关的资金信息和披露(如果有的话)将在文章末尾提供。
文章加工费由Grifols, S.A.资助
提交并经外部同行评审。处理编辑是Anthony Amato,医学博士,FAAN。
证据类别:NPub.org/coe
信息图表NPub.org/ig1007
- 收到了2022年1月31日。
- 最终接受2022年9月16日。
- ©2022作者。由Wolters Kluwer健康公司代表美国神经病学学会出版。首页
这是一篇开放获取的文章,根据创作共用署名-非商业性-非衍生品许可4.0 (CC BY-NC-ND),该网站允许下载和分享论文,前提是论文被正确引用。未经本刊许可,不得以任何方式更改作品或将其用于商业用途。
参考文献
信件:快速在线通信
需求
如果你要上传关于文章的信件:
您必须在六个月内更新您的披露:http://submit.首页neurology.org
您的合著者必须发送一份完整的出版协议表格来首页(对于主要/通讯作者不需要填写以下表格即可),然后再上传您的评论。
如果你在回复一篇关于你最初撰写的文章的评论:
您(和共同作者)不需要填写表格或检查披露,因为作者表格仍然有效
并适用于信件。
提交规格:
- 文章必须少于200字,参考文献少于5篇。参考文献1必须是你所评论的文章。
- 投稿者不得超过5人。(例外:原作者回复可以包括文章的所有原作者)
- 只可提交发稿日起6个月内发表的文章。
- 不要冗余。在提交之前阅读文章上已经发布的任何评论。
- 提交的意见在发表前须经过编辑和编辑审查。

{kind=link}
{kind=link}
{kind=link}
{kind=link}